Beware of confounding basecaller versions
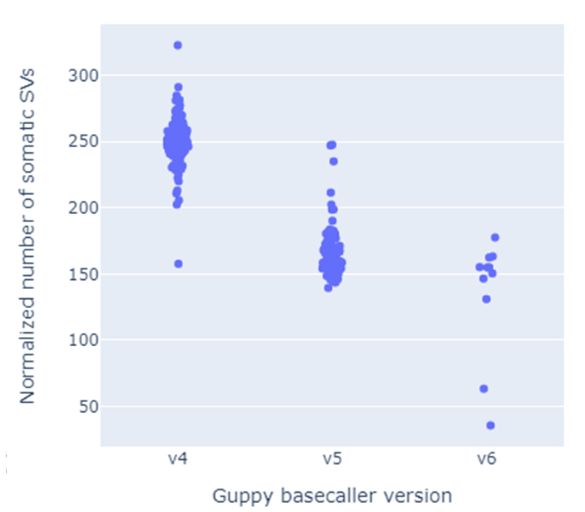
Of course, having a newer version of the nanopore base caller will have some impact. The accuracy obviously constantly improves, and in long-running projects like ours, we now have samples with Guppy4, Guppy 5, and Guppy 6 base caller versions.I reasoned that it would probably be a confounder, but we mainly look at structural variation. Slight changes in nucleotide level accuracy could change the breakpoints by a bit. However, the bigger-picture variant would still be the same, especially as this combines evidence from multiple reads.However, that last part is specifically where things start breaking down. If you look at germline structural variants, where multiple reads are combined, all seems fine. Combining basecaller versions could be better, but okay. Nevertheless, more recently, I looked at somatic variants, where I trusted any variant as soon as it had the support of only two reads. Below I summarize the number of somatic variants (normalized to the median genome coverage) and stratify the datasets per major version of the basecaller. Since that shows quite a drastic effect, I am now synchronizing the entire cohort to the same version of Guppy 6, involving moving around 400 terabytes of data and heavily occupied GPUs.