A brief follow-up on SVIM
Wouter De Coster
long readsna19240structural variants
300 Words · 1 Minute, 21 Seconds
2019-04-08 15:27
In a previous post I looked at SVIM, a new structural variant caller intended for long reads. Since then some changes have been done, and today I evaluated v0.5.0 using our PromethION data from NA19240, base called using Guppy flipflop. As recommended by the author I filtered on a quality score of 10. (Note that in the previous version it was recommended to use a quality filter of 40.) SVIM is pretty fast, and I’m also happy with its accuracy. The evaluation I did is genome-wide and on GRCh38. For a variant to be considered concordant (true positive) compared to the truth set it should have:
the same type (DEL/DUP/INV)the pairwise distance between the breakpoints should be maximally 500 bp
Other evaluations exclude some parts of the genome which are more challenging, such as segmental duplications (but that’s just where things get interesting), allow a larger distance between breakpoints or require a certain percentage reciprocal overlap between SVs.
The precision I got was 0.698 and the recall 0.714, together giving an F-measure of 0.706, which is decent, compared to other SV callers. Sniffles is more precise, but less sensitive. The future of SV calling looks bright!
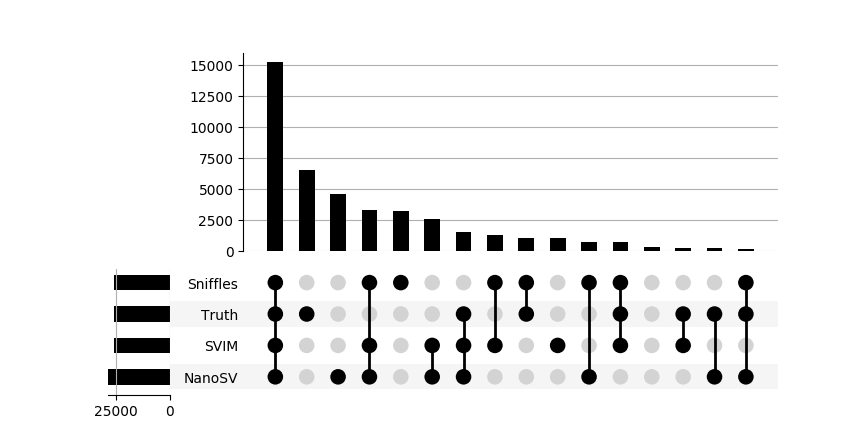
For merging SVs in my evaluation I use SURVIVOR, which I have wrapped in a Python tool which I called surpyvor. It has reasonable default settings for parameters of SV merging in SURVIVOR, and a more convenient command-line interface. In addition you can use the highsens and highconf sub-commands to create a high-sensitivity SV set (taking the union from the input) or to create a high-confidence SV set (taking the intersection from the input). I also included sub-commands to create a Venn or Upset plot, of which the latter is a great way to show proportional overlaps between sets (see below for an example).