Insertion-deletion ratio in Oxford Nanopore PromethION data
It’s been a while since I wrote another blog post, so here is something short. Today I had to figure out the inversion deletion ratio in Oxford Nanopore Sequencing data from PromethION human whole genome sequencing.
I want to compare this ratio for reads which are qualified by albacore as “pass” (average base call quality score above 7) and “fail”(average base call quality score below 7). For speeding things up, I only used chromosome 22 for this. samtools view anon_pass.bam chr22 -o anon_pass_chr22.bam samtools view anon_fail.bam chr22 -o anon_fail_chr22.bam To get the insertion-deletion ratio I will use the CIGAR string from the aligned reads. The Python script can be found below. The code uses pysam and iterates over the bam file, using the cigartuples attribute. In this attribute, insertions get a “1” and deletions a “2"as the cigar operation code (op). The other part of the tuple is the length of the operation. So a tuple of (1,25) corresponds to an insertion of 25 nucleotides relative to the reference genome.
For deletions, the minimum is 1 to avoid divide by zero errors. The boolean expression on line 18 will evaluate to 1 if the first part evaluates to 0.
https://gist.github.com/wdecoster/a2be73a676b68a9f92d07509e30d37a1
From these ratios I create a simple histogram using matplotlib, using bins of 0.1 with a maximum of 5. The resulting plots are shown below. So the ratios are:
pass: 1.002 fail: 1.027 combined: 1.012
Indeed showing a difference between failed and passed reads. That’s all for today!
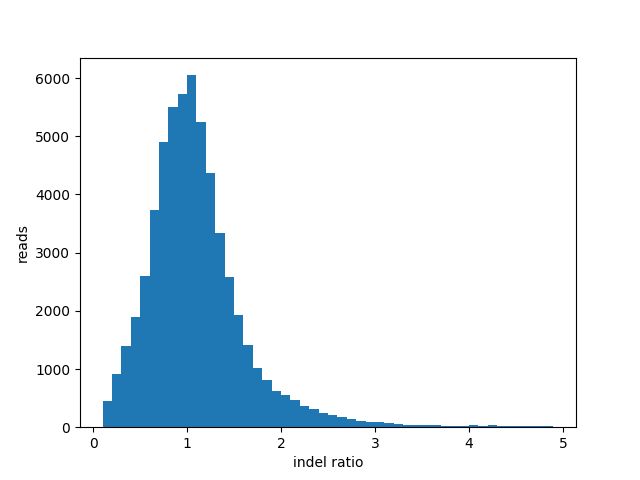 Insertion Deletion ratio for “fail” reads
Insertion Deletion ratio for “fail” reads
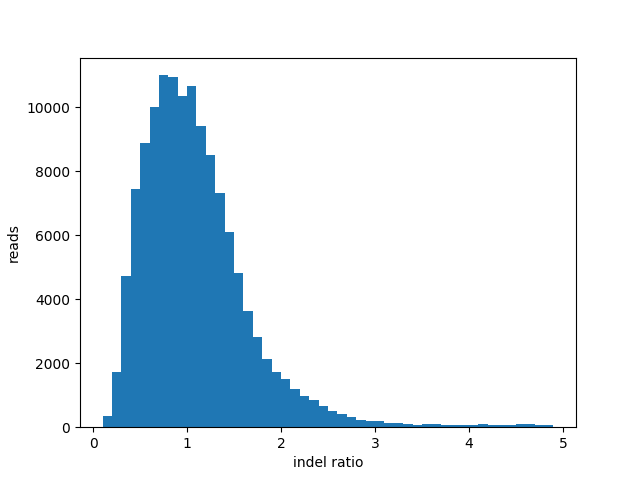 Insertion Deletion ratio for “pass” reads
Insertion Deletion ratio for “pass” reads